
BioStock’s article series on drug development: The Success of Orphan Drug Designation
Getting a pharmaceutical drug approved by regulators is notably very challenging, requiring several years and exorbitant amounts of money. However, recent studies following trends in drug development approval rates tell a heart-warming story of approval rates going up in the last decade, especially when a drug reaches phase III clinical trials. The data should get investors betting on one class of drugs in particular – orphan drugs. With this fifth and final piece, we round out this series on drug development by taking a closer look at orphan drug designation and its benefits (as well as some of its flaws).
In its complexity, one thing is clear – a drug’s path to market approval is swamped with obstacles, mainly tough regulations aimed at following pharmacovigilance guidelines that slow down the drug development process and lead to low rates or approval.
Despite the negative impactive some of the regulations may have on drug development, their purpose is essential – keeping patients safe from potential toxic side-effects. So, generally speaking, regulators remain firm in their strict stance, even as they are being hard-pressed to be more flexible. However, in recent years, the regulating authorities have been listening to drug makers and patients and have created new rules to help some new drugs have a higher chance of reaching the market, especially those developed for serious illnesses with unmet medical needs. We took a closer look at those new rules in part IV (add link) of this series and we briefly mentioned the Orphan Drug Act as an important example.
However, the implications of that particular bill require further analysis, which we provide here.
The Orphan Drug Act
The Orphan Drug Act, passed in 1983 by the FDA to facilitate the development of drugs targeting rare diseases, or, as by the FDA’s definition, diseases that affect fewer than 200,000 people at any given time. The act came to fruition as patients with rare diseases felt increasingly ignored by drug companies, thus the name orphan drugs.
It paved the way for three major economic incentives for drug companies developing drugs able to receive orphan drug designation (ODD) from the FDA. The major one is market exclusivity – no direct competition from other companies for 7 years when treating a rare disease. Other incentives include tax credits and clinical research subsidies.
In 2000 the EU followed in those footsteps by creating the Committee for Orphan Medicinal Products (COMP), which gives pharma companies similar incentives, including market exclusivity. There are however some major differences compared to the FDA. First, COMP gives 10 years of market exclusivity instead of 7; moreover, it requires the drugs to be for serious medical conditions and for which there is currently no alternative already on the market – something that is not enforced by the FDA, thus making it easier to receive orphan designation in the US.
An increase in drug approvals
The benefits of the bill have been quite evident. Since its passage, the number of medications approved for rare diseases has skyrocket from just 34 in 1983 to more than 600 today, reaching millions of people.
Over the last 5-10 years, however, there is substantial evidence suggesting that ODD has led to major benefits in the drug development process as well. As has been reported by Monocl Strategy and Communication earlier this year, recent studies have shown that phase III drug approval success rates have increased substantially over the last decade, and that orphan drugs may be to thank for pushing those rates higher.
The main results from one of the studies in question, published this year in Nature Reviews, are summarized in an article by Endpoints News, which details success rates of drugs moving from phase III to launch rising from 49 per cent to 62 per cent between 2010 and 2018. The study points out that success rates for orphan drugs have surged the most within that period (see graph).
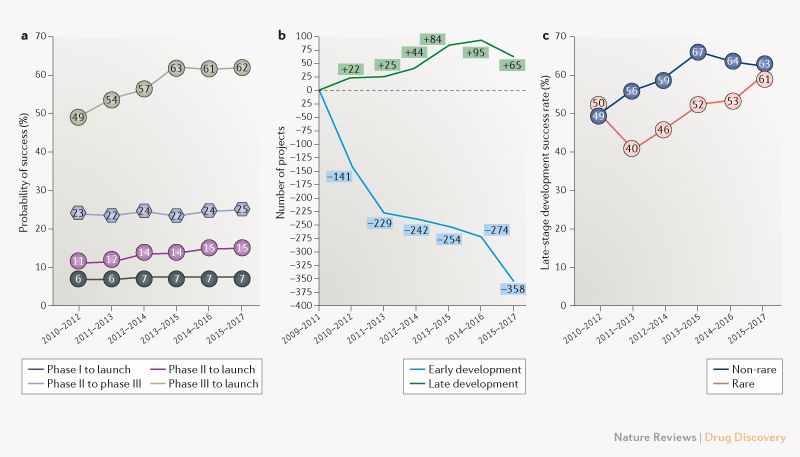
Those results run in parallel with data from a study by BIO, which suggests that the average success rate of orphan drugs in clinical development was greater than that of all drugs pooled together between 2006 and 2015. Non-oncology orphan drugs in particular had a probability of success (POS) of reaching approval from phase I of 25 per cent. Compare that to all drugs averaged together with 10 per cent POS.
That particular study decided to separate oncology drugs from orphan drugs, although in many cases they overlap – something to keep in mind since oncology drugs trend quite low in terms of POS. More on that in the next section.
Success for orphan drugs is recent
It’s not all roses for orphan drugs. A study by MIT published last year tells a slightly different story when looking at a wide range of clinical trial data from 2000 to 2015. Within that time frame, orphan drugs had one of the lowest average probability of success (POS) rates (6.2 percent) to reach approval from phase I, and an average phase III POS rate of 46.7 per cent, well below the 59 per cent POS rate encompassing all drugs. Those percentages cover all orphan-designated drugs, including those for treating cancer, which, as mentioned above, brings down the POS averages.
Of course, one should keep in mind that the different results from these three studies come from analyses of three different time frames. However, the common thread among the studies is that they show a dramatic rise in drug approvals from 2013, especially among orphan drugs. This is particularly apparent in the Nature Reviews study, which has a time frame that extends to 2018, making it slightly more relevant for predictions going forward, since the other studies only extend to 2015.
Orphan drugs require post-approval studies
A relevant factor to consider when discussing orphan drug success rates and market approval is that, more often than not, orphan drugs have to go through stringent post-approval safety testing. This is because knowledge about rare diseases is still scarce (since they are rare), so patients need to be followed for several years to be sure a drug is safe and effective.
Moreover, rare diseases predominantly affect children or young adults, which adds to the need for post-approval clinical trials, since orphan drugs are typically tested in adults for approval. This creates an impasse for recruiting clinical trial volunteers and adds complexity to clinical trial design. Clinical trial recruitment is further complicated by the fact that rare diseases involve a much smaller number of affected patients, thus adding to the difficulty in finding an adequate number of clinical trial volunteers.
Overall, post-approval studies are a fundamental aspect of orphan drug clinical trial design made to address these and other limitations.
With success comes scepticism
Despite the positive drug development trends for drugs receiving ODD, the Orphan Drug Act has received its healthy share of criticism. For one, reports indicate that still 95 per cent of the 7000 documented rare diseases still lack treatment options, yet, this class of drugs accounted for a majority of FDA approvals in 2018 and is one of the most profitable raking in close to 140 BUSD from sales last year.
Furthermore, an investigation by Kaiser Health News shows that drug developers take advantage of loopholes in the Orphan Drug Act to maximise their profits. In fact, the investigation revealed that several mainstream drugs were later “repurposed” to treat rare diseases in order to receive ODD, thus leading to a monopoly in the orphan drug market thanks to market exclusivity granted from the legislation.
A good example is AbbVie’s Humira (adalimumab), which happens to be the top-selling drug in the world with revenue from sales reaching 13 BUSD annually. The drug was originally approved for a non-orphan indication (rheumatoid arthritis), but now has 12 indications, six of which have orphan designation. As a report undertaken independently by the QuintilesIMS Institute shows, the orphan indications amount to 3.8 per cent of the annual sales volume generated by Humira.
This leads to speculation that the Orphan Drug Act isn’t necessarily doing what it was intended to do – create treatments for patients with urgent unmet needs within rare diseases.
All said and done, despite the criticism, there is plenty of evidence showing that the Orphan Drug Act has done a lot to encourage the development of drugs for rare diseases and has helped increase the development success rates within pharmaceutical development, especially within the last decade.
This concludes our article series on drug development. If you missed our previous articles, you can find them at these links.
Part I: The Drug Development Process
Part II: Challenges Associated with Drug Development
Part III: A Brief History of Regulatory Affairs
Part IV: Making Treatments More Accessible to Patients



