
regulatory framework, IVDR, is replacing the old directive, IVDD. The anticipated changes in the new regulation are likely to create a complicated and time-consuming transition process for many of the manufacturers of in vitro diagnostic medical devices. In this article, BioStock presents some of the key changes in IVDR compared to the current directive.
In vitro diagnostic medical devices (IVDs) are used for the examination of specimens, including blood and tissue donations, derived from the human body to determine the status of a person’s health. A few examples are blood glucose monitors, pregnancy tests and products for the detection of infections.
IVDR will fully apply in 2022
The In Vitro Diagnostic Regulation (IVDR) entered into force in 2017, marking the start of a five-year transition period for IVD manufacturers. IVDR will fully apply in all EU member states from 26 May 2022. After this date, all manufacturers of IVDs (except for those with valid certificates issued by an EU notified Body) need to be conforming to the requirements in IVDR to be able to place their in vitro diagnostic medical devices on the EU market.
The transition deadline may seem far away, but a number of activities should already be underway to make sure that the devices meet the requirements in IVDR before the end of the transition period.
IVDR is an improved version of the current directive
The new regulation will replace the directive on in vitro diagnostic medical devices (IVDD), which has been the regulatory framework for placing IVDs on the EU market for over 20 years. The IVDR is immediately binding for all EU countries without need to be transferred into the law of each state, which was the case with the IVDD.
IVDR will ensure the safety and performance of all in vitro diagnostic medical devices in Europe. Overall, the new regulation represents a significant strengthening of the existing regulatory system. Several requirements in IVDR are the same as in the old directive, but there are also some major differences between the two.
Expanded scope
Under IVDR, the product scope will be expanded to cover more types of devices, for example genetic tests and other predictive tests that provide information about a patient’s predisposition to a medical condition or a disease. Also added to the scope are products that predict treatment response or reactions, e.g., companion diagnostics that are co-developed with drugs to make sure that the treatment is safe and effective.
Classification changes dramatically
One of the biggest changes introduced by the IVDR is a new rule-based classification system of four risk categories, ranging from low risk (class A) to high risk (Class D).
Many IVD devices are currently self-declared under the IVDD, which means that Notified Body involvement is not required. However, this will change drastically with the introduction of the IVDR. The majority of IVDs will move from being self-declared under the IVDD, to requiring Notified Body assessment under the IVDR.
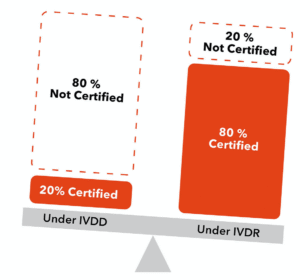
80 per cent will need Notified Body certification
About 80 per cent of IVDs are expected to fall into risk classes B, C or D under IVDR, all of which require some level of Notified Body oversight. This means that several IVD manufacturers will now require Notified Body conformity assessment to keep/put their products on the market, which increases the regulatory burden for both manufacturers and their Notified Bodies.
Consequently, the workload for Notified Bodies will increase due to the stricter requirements and a higher level of oversight from the Notified Bodies. The next article in this article series will address the fact that Notified Bodies may become regulatory bottlenecks in the shift from the old directives to the new regulations.

Tina Amini comments
BioStock has reached out to Tina Amini, Director of Medical Device Division at NDA Group, for her expert input and knowledge within the field. Apart from a new rule-based classification system and an increased level of Notified Body oversight, IVDR leads to several changes according to Tina Amini:
- Introduction of EU Reference Laboratories (EURLs). These laboratories have a key role in verifying the safety and performance of IVDs both in the pre-market and post-market lifecycle of a device.
- The administrative burden will increase substantially as a result of registration requirements and the transparency measures that build on the Unique Device Identification (UDI).
- More focus on risk management.
- More detailed information concerning the analytical and clinical performance of the device.
- Introduction of the Scientific Validity concept. Manufacturers will need to demonstrate scientific validity through literature reviews, expert opinions or proof of concept studies/clinical performance studies.
- With the requirement for Post-Market Performance Follow-up, the scope for Post-Market Surveillance has been broadened. As a result, class C and D IVDs must have an annual update as part of a Periodic Safety Update Report (PSUR).
- A lifetime approach.
Improved quality and safety
In general, quality, safety and reliability of IVDs will be improved by the IVDR, especially since IVDR expands the Post Market Surveillance activity requirements. Under the new regulation, IVD manufactures will be required to proactively and systematically gather and analyze relevant data throughout the lifetime of the devices. This leads to earlier detection of potential product malfunctions and ways to improve the devices that are put on the EU market.
This was the third article in BioStock’s article series about the major shift in the regulatory landscape for medical devices. Read the other articles in the article series:
