
Läkemedelsutveckling – De fyra stegen
Att utveckla ett nytt läkemedel är en komplicerad, mångfacetterad och lång process – vanligtvis tar det minst ett decennium. Varje utvecklingssteg är dessutom förknippat med nya utmaningar för det läkemedelsutvecklande bolaget. I den här artikelserien går BioStock igenom läkemedelsutvecklingens många aspekter och vad som krävs för att ta ett läkemedel hela vägen till marknaden.
De flesta människorna har någon form av relation till läkemedel. Dock tänker vi sällan på arbetet som ligger bakom utvecklandet av dessa. I den här artikeln tittar BioStock närmare på de fyra stadierna av läkemedelsutveckling, inklusive några av de viktigaste regulatoriska hinder som läkemedelsutvecklare möter på vägen mot att kunna förse patienterna med ett nytt läkemedel.
Läs även:
Läkemedelsutveckling del II – Utmaningarna
Läkemedelsutveckling del III – En regulatorisk hinderbana
Läkemedelsutveckling del IV – Att göra behandlingar tillgängliga
Läkemedelsutveckling del V – Framgången med särläkemedel
Läkemedelsutveckling tar ofta mer än ett decennium
Att utveckla nya, innovativa läkemedel är en lång process. Den genomsnittliga tiden från upptäckt till marknaden är 12 år, medan det inom nya medicinska områden kan ta uppemot 30 år. Många aktörer skulle nog anse att 12 år är en optimistisk uppskattning, inte minst med tanke på att endast 1 av 5000 nya substanser till slut godkänns för försäljning av tillsynsmyndigheter som Food and Drug Administration (FDA) i USA eller European Medicines Agency (EMA) i EU.
Läkemedelsutvecklingens fyra steg
Läkemedelsutveckling kan delas upp i fyra steg: upptäckt, prekliniska studier, klinisk utveckling och marknadsgodkännande. Bilden nedan ger en översikt av processen med en uppskattad tidslinje för respektive steg.
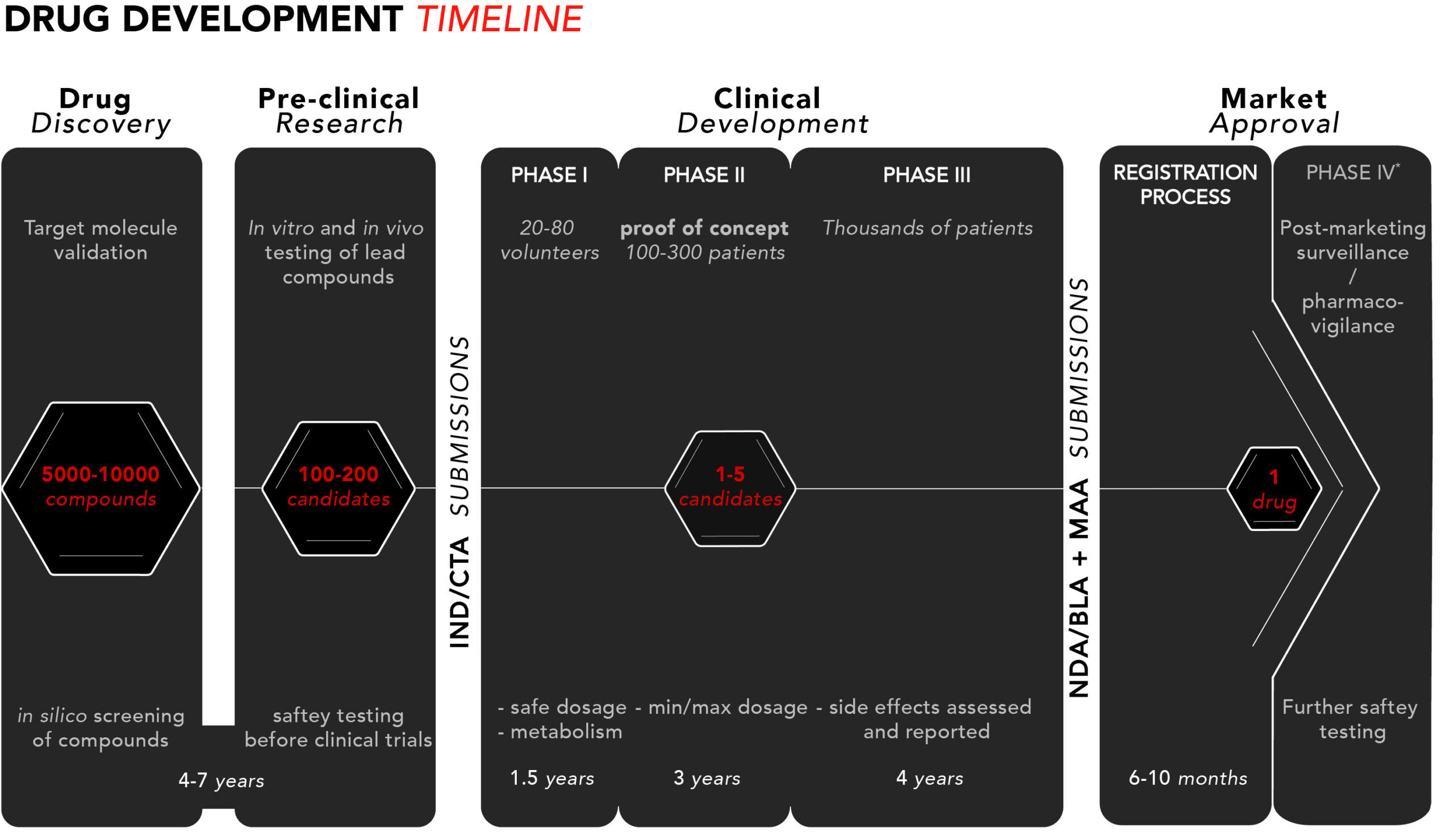
Låt oss gå igenom processen steg för steg.
1. Upptäckt
Upptäcktsstadiet börjar med forskning och utveckling i laboratoriemiljö. Forskare identifierar målmolekyler – t.ex. gener eller proteiner – som tros ha potential att bidra till att en sjukdom utvecklas eller förvärras.
Därefter utförs så kallade in silico-tester av hundratals – eller tusentals – kemiska eller biologiska föreningar för att utvärdera dess förmåga att interagera med den identifierade målmolekylen. Detta görs i syfte att hitta den förening som har bäst förutsättningar att påverka sjukdomen.
2. Preklinisk forskning
Efter att ha valt ut en lämplig förening följer den prekliniska forskningen. Föreningen byter namn till ”kandidaten” och denna testas i två experimentella modeller: in vitro och in vivo. In vitro är latin för ’i glas’, vilket innebär att kandidaten testas på celler i provrör eller andra behållare. In vivo betyder ’i livet’, och i dessa tester så utvärderas kandidaten i djurmodeller.
Under den prekliniska fasen får forskarna en första uppfattning om hur kandidaten kan interagera i människokroppen. Den viktigaste aspekten av preklinisk forskning är de rigorösa säkerhetstester vars syfte är att se till att kandidaten inte är giftig innan den kan testas i kliniska studier på människor.
Sammantaget kan upptäcktsfasen och den prekliniska fasen ta fyra till sju år.
Efter färdigställande av de prekliniska testerna så är det – givet goda prekliniska resultat – dags att ansöka om tillstånd för att gå vidare till kliniska studier. Detta görs antingen via ett IND-program (Investigational New Drug) i USA eller en klinisk prövningsansökan (Clinical Trial Application, CTA) i EU. Respektive tillsynsmyndighet inspekterar då all tillgängliga prekliniska data och beslutar om huruvida kandidaten är redo för kliniska studier.
3. Klinisk utveckling
Fas I – säkerhet
Efter myndighetsgodkännande samt godkännande från etiska kommittéer inleds den första kliniska studien, en fas I-studie – som utgör den första studien i människa. Här testas kandidaten i allmänhet på 20 till 80 friska frivilliga individer med syfte att avgöra om kandidaten beter sig på samma sätt i den mänskliga kroppen som de prekliniska studierna har indikerat.
Substansens säkerhetsprofil – eller toxicitet – är återigen huvudfokus, men den här gången i människa. Man testar vad som utgör en säker dos, hur läkemedlet tas upp i kroppen, och hur länge det är aktivt i kroppen. Det är värt att notera att kliniska fas I-studier av säkerhetsskäl tenderar att utesluta kvinnor i fertil ålder.
En fas I-studie tar upp till ett år att genomföra.
Fas II – Proof-of-Concept
Vid goda säkerhetsresultat från fas I ansöker man om tillstånd för nästa kliniska utvecklingssteg – fas II. Här testas kandidaten oftast i mellan 100 och 300 patienter som diagnosticerats med den sjukdom som kandidaten är avsedd att behandla. Utöver att fortsätta utvärdera säkerhetsparametrar läggs nu även effektmått till raden av utfallsmått (eng: endpoints). Därutöver ska fas II-studien också fastställa minsta och maximala dos, med fokus på att kunna bestämma de doser som ska användas i nästa fas.
Fas II tar i genomsnitt två år.
Fas III – regulatoriska bevis
Vid goda säkerhets- och effektdata från fas II ansöker man om tillstånd för fas III. Antalet patienter som ingår i en fas III-studie är vanligtvis minst 1000, i syfte att få in tillräckligt med data för att visa att kandidaten beter sig som önskat – både avseende säkerhet och klinisk effekt.
I samband med fas III-studien dokumenteras och rapporteras också alla eventuella biverkningar som patienterna upplever, därför måste studiepersonerna exponeras för kandidaten under en lång tidsperiod för att säkerställa att alla biverkningar bedöms ordentligt. Biverkningarna som noteras i detta skede är de som sedan listas i slutproduktens bipacksedel.
Fas III tar i genomsnitt ett till fyra år.
4. Marknadsgodkännande & lansering
Registreringsprocessen
Vid goda resultat från fas I-III lämnar man in en ansökan för marknadsgodkännande, kallad New Drug Application (NDA)*/Biologics License Application (BLA) i USA och Marketing Authorisation Application (MAA) i EU. Dessa kan omfatta 100 000-tals sidor med dokumentation som sammanfattar all insamlade data från upptäcktsfasen och framåt, och där huvudmannen argumenterar för ett godkännande hos FDA och/eller EMA.
*Notera att vissa nya läkemedel kategoriseras som new molecular entities (NMEs), alltså aktiva ingredienser som ännu inte godkänts av FDA eller andra tillsynsmyndigheter, och att dessa kräver en NME NDA-ansökan.
Att förbereda ansökningsdokumentationen kan ta flera månader, följt av cirka 6–10 månader för myndigheterna att behandla ansökan.
Marknadslansering
Om läkemedelsmyndigheterna godkänner en ansökan är kandidaten – eller läkemedlet som det nu kallas – redo för marknadslansering. Vid denna tidpunkt börjar prisförhandlingar mellan huvudmannen och de potentiella köparna (myndigheter eller försäkringsbolag, beroende på vårdsystem).
Prisförhandlingsprocessen kan skilja sig mycket åt från land till land. EU:s medlemsstater följer specifika EU-riktlinjer men kan därtill ha vissa landspecifika riktlinjer. I t.ex. Sverige fastställs ersättningsbestämmelserna på nationell nivå. Läs mer. I USA, däremot, sköts prisförhandlingarna mellan läkemedelsbolaget och de privata försäkringsbolagen, utan inblandning från myndigheter. Systemet har lett till betydligt högre priser på läkemedel i USA jämfört med Europa och andra utvecklade länder.
Fas IV-studier – övervakning av marknadsföring och säkerhet
I vissa fall kräver tillsynsmyndigheterna uppföljande fas IV-studier efter det att ett läkemedel erhållit marknadsgodkännande. Detta görs genom att samla in data från klinisk praktik – alltså verkliga vårdenheter som behandlar patienter.
Syftet är en utökad säkerhetsövervakning av läkemedlet (s.k. farmakovigilans). Fas IV-studier utvärderar dels huruvida läkemedlet påverkas av andra läkemedel, och dels genomförs ytterligare säkerhetstester. Detta är inte minst viktigt när det gäller läkemedel för komplexa medicinska tillstånd, eller som avser behandla gravida kvinnor som sannolikt inte har varit inkluderade i studierna i fas I-III.
Därutöver kan fas IV-studier vara relevanta för läkemedel som ska behandla sällsynta tillstånd, som ofta innebär begränsat antal patienter i fas I-III. Resultaten från de tidigare kliniska studierna har därmed en svagare statistisk säkerhet, varför myndigheterna efterfrågar ytterligare bekräftelse på läkemedlets säkerhet och effekt.
Artikelseriens kommande delar
Sammantaget tar det alltså 12–15 år från substansupptäckt till dess att ett läkemedel når marknaden. Notera att vi i denna artikel inte har berört de utmaningar som är kopplade till varje enskilt utvecklingssteg och myndighetsbesked. Håll utkik efter nästa artikel i serien där vi tittar närmare på de utmaningar som läkemedelsutvecklande bolag står inför, inklusive de höga kostnader som läkemedelsutveckling är förknippat med.



