
BioStock’s article series on MDR and IVDR: An introduction to the upcoming changes
The regulatory landscape in Europe for medical devices is changing drastically as two new regulations, MDR and IVDR, are being implemented. The date for implementation of MDR is now less than six months away, but many of the companies are not ready to meet the new and stricter requirements. Some manufacturers may even have to withdraw their medical devices if they do not meet the requirements on time.
This is the first of four articles in which BioStock takes a closer look at the major shift in the regulatory landscape for medical devices. In this article, we will go through the key deadline dates as well as other useful information concerning the regulations.
 BioStock has also had the opportunity to talk to Tina Amini, Director of Medical Device Division at NDA Group, a world leading regulatory, drug development and medical device consultancy. Tina Amini’s role at NDA is to lead the team in providing support for MedTech and Pharma companies regarding their medical devices and in vitro diagnostic devices (IVD), including companion diagnostics and combination products.
BioStock has also had the opportunity to talk to Tina Amini, Director of Medical Device Division at NDA Group, a world leading regulatory, drug development and medical device consultancy. Tina Amini’s role at NDA is to lead the team in providing support for MedTech and Pharma companies regarding their medical devices and in vitro diagnostic devices (IVD), including companion diagnostics and combination products.
The current directives are outdated
The current directives – MDD, AIMDD and IVDD – have been the official legislative acts for medical devices in Europe for over 25 years. During this period, a number of scientific and technological advances have been made and several shortcomings in the directives have been identified.
Thus, the European Commission decided to adopt two new EU regulations in 2017 – the Medical Devices Regulation (MDR) and the In Vitro Diagnostic Medical Devices Directive (IVDR). These two regulations will give Europe a more up-to-date and internationally applicable regulatory framework that is more in line with regulations for medical devices in the rest of the world.
Are manufacturers ready for this major shift?

MDR will be fully applied in less than six months (26 May 2021) in all EU member states, while the deadline to comply with IVDR is set to 26 May 2022. All stakeholders in the sector are facing several challenges in the preparations for the new regulations, not least the manufacturers of medical devices and in vitro diagnostic medical devices.
BioStock reached out to Tina Amini to get her take on whether or not the manufactures are prepared for the transition:
»Meeting the new and stricter requirements, putting in place processes to meet the requirements and updating technical files requires knowledge, time and resources and of course the finances. Not all have the in-house capabilities/resources/funds so there is a risk of not being able to continue marketing the devices in the EU if they do not meet the requirements on time« – Tina Amini
NDA supports the manufacturers in the transition work
NDA provides a full service from initial concept to post market. The support can involve providing training to manufacturers on the new and expanded requirements in the regulations or implementing the requirements. The latter can be achieved via an initial gap analysis of their current QMS and technical files to compare them against the new requirements in order to identify gaps, and then preparing a remediation strategy for the company.
Before a medical device can enter the EU market, it usually needs to be assessed by a Notified Body (NB) in order to obtain a CE certificate. For those companies that do not have a NB or are looking for a new NB designated under MDR/IVDR, NDA provide support in selection and subsequent interactions with the selected notified body.
Three EU directives become two regulations
“MDR replaces MDD” is the most commonly used description of the ongoing change in the regulatory landscape for medical devices, but in actual fact, three EU directives are replaced by two EU regulations.
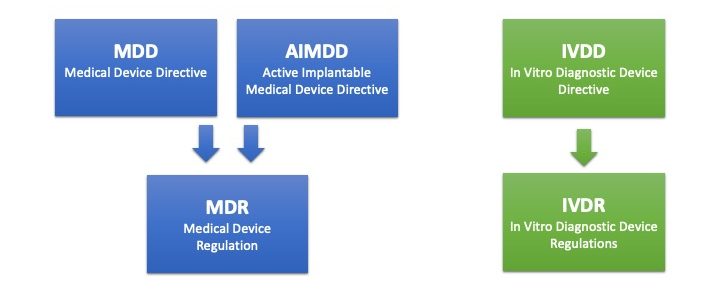
The Medical Device Directive (MDD) and the Active Implantable Medical Device Directive (AIMDD) are merged into one legislation – the Medical Device Regulation (MDR). The In Vitro Diagnostic Medical Devices Directive (IVDD) is in turn replaced by the In Vitro Diagnostic Medical Devices Regulation (IVDR).
The important difference between directives and regulations
It is important to note that the directives are being replaced by regulations. Regulations become automatically binding in all EU member states on the date that they come into effect, while directives must be incorporated into national law.
With the old directives for medical devices, each EU member state interpreted and applied the legislation in its own way, but with MDR and IVDR, the same legislation will be applied in all member states. This creates a greater legal certainty and a more uniform market for medical devices within the EU.
Deadline postponed due to Covid-19
A medical device may be placed on the market within the EU only if it complies with the new regulation, MDR. However, the medical device manufacturers have been given a transition period in which to undertake the extensive work necessary to meet the requirements set by the new regulations.
The transition period for MDR was originally supposed to be three years – until 26 May 2020 – but due to the global outbreak of Covid-19, it was extended by one year in order to secure the supply of medical devices during the pandemic. On and after 26 May 2021, in order to access EU market, “new” medical devices need to conform to the requirements of MDR.
The manufacturers for in vitro diagnostic devices (IVDs) were given a five-year transition period, which means that 26 May 2022 is the deadline for complying with IVDR.
A grace period
The manufacturers that need more time to meet the requirements in the new regulations actually have a little bit more breathing space after the original deadlines. Medical devices with a certification under MDD/AIMDD/IVDD are allowed to utilise another transition period, also called a “grace period,” that lasts until 26 May 2024.
This means that some certificates according to the old directives are still valid until 26 May 2024 – after this date, these devices cannot be placed on the market. According to Tina Amini, most of the companies have chosen to extend their certificates according to the old directives:
»Most manufacturers have opted to renew their MDD/AIMD certificates to benefit from the transitional period. This could be either because they need more time to comply with MDR or they need further clarification/guidance on the requirements.«
A renewal of the CE mark in compliance with the old directives is a possible route to take in order to gain some time. However, the grace period only applies under certain conditions. For example, no significant changes in the product’s design or intended purpose are allowed to be made during the period. This means that the device innovation is essentially “frozen,” which affects the company’s competitive position on the market.
Additionally, there are some requirements in MDR that must be applied by all manufacturers after 26 May 2021. Examples of such requirements relate to post-market surveillance (PMS), market surveillance, vigilance, registration of economic operators and of devices. This also applies to the manufactures that decided to extend their old certificates.
See the most important deadline dates, according to Tina Amini, below.
[divider]KEY DATES[/divider]
|
26 May 2021
|
In order to access the EU market, “new” medical devices need to be conforming to the requirements of MDR by this date.
After 26 May 2021, devices with valid MDD/AIMD certificates need to meet MDR requirements relating to post-market surveillance (PMS), market surveillance, vigilance, registration of economic operators and of devices. |
|
26 May 2022
|
Deadline for in vitro diagnostic devices to comply with IVD regulations, with the exception of the IVDs with valid NB certificates issued under IVDD
|
|
26 May 2024
|
Deadline for medical devices with valid MDD/AIMDD/IVDD certificates issued by a NB to be placed on the market/- Directive certificates become void.
|
|
26 May 2024
|
Deadline for Class I devices (self-certified under MDD/AIMDD) which under MDR are up-classified and require NB involvement
|
|
26 May 2025
|
Last date for putting into service / making available devices certified under MDD/AIMDD/IVDD
|
The countdown has begun
The transition to MDR and IVDR will be challenging for all involved. Comprehensive work is required by all companies within the industry, Notified Bodies, and the European Commission.
The companies should have begun the transition work a long time ago, preferably already when the new regulations came into force in 2017. It is not unlikely that we will see medical devices being withdrawn from the market meaning that MDR and IVDR will have a significant impact on the competitive landscape for medical devices in the future.
Having clinical evidence will be more important now when the new regulations are introduced, and generating such data is easier said than done – it can take several years and requires a lot of resources. BioStock will look at what other requirements are being introduced in MDR and IVDR in subsequent articles of this series.
Part I: An introduction to the upcoming changes
Part IV: Notified Bodies – potential bottleneck



