
RhoVac utvecklar immunterapeutiska läkemedel och huvudkandidaten RV001 är ett terapeutiskt cancervaccin med potential att förhindra eller begränsa metastasering vid cancer. Kandidaten har tidigare visat lovande resultat i en fas I/II-studie inom prostatacancer, vars primära syfte var att utvärdera kandidatens säkerhetsprofil och tolerabilitet. Ett förmånligt utfall avseende dessa parametrar kunde konstateras under hösten 2018, men inte nog med det. RhoVac kunde dessutom fastställa att 18 av 21 patienter (86 procent) uppvisade ett robust RV001-medierat immunsvar, klassificerat som Confirmed Immune Response.
I januari i år kunde bolaget vidare meddela att samtliga 18 patienter med konstaterat immunsvar uppvisade ett likvärdigt vaccinmedierat immunsvar även vid 3- respektive 6 månadsuppföljningarna. Slutliga uppföljningsresultat, som även kommer att inkludera uppföljningar efter 9 och 12 månader, förväntas i mitten av 2019.
Efter framgångarna i fas I/II planerar RhoVac nu en klinisk fas IIb-studie i prostatacancerpatienter i tidigt sjukdomsskede som ska inledas i mitten av 2019.
Vill genomföra studie även i USA
RV001 har hittills utvecklats och studerats i Europa – då främst vid universitetssjukhuset i Köpenhamn, Danmark, och Tübingen Universitet, Tyskland – men RhoVac undersöker nu möjligheten att förlägga delar av den fortsatta kliniska utvecklingen i USA. Anledningen är att man vill kratta manegen för en framtida introduktion även på den amerikanska marknaden.
Som ett led i sitt geografiska avancemang ansökte RhoVac tidigare i år om ett pre-IND-möte med den amerikanska läkemedelsmyndigheten FDA. Ett möte som myndigheten nu har bekräftat.
Viktigt att samverka med regulatoriska myndigheter
För läkemedelsutvecklande bolag går vägen till patienten alltid via de regulatoriska myndigheterna som har i uppgift att se till att patienter får tillgång till effektiva och säkra behandlingar så snabbt som möjligt. Genom att tidigt engagera de regulatoriska myndigheterna, och därigenom undvika onödiga fallgropar i sin utvecklingsprocess, kan läkemedelsutvecklande bolag förkorta sin väg till marknaden.
För att få starta en klinisk studie krävs tillstånd från de regulatoriska myndigheterna i de länder där den kliniska studien kommer att genomföras. Ansökan om tillstånd för klinisk prövning kallas CTA (Clinical Trial Application) i EU och IND (Investigational New Drug application) i USA.
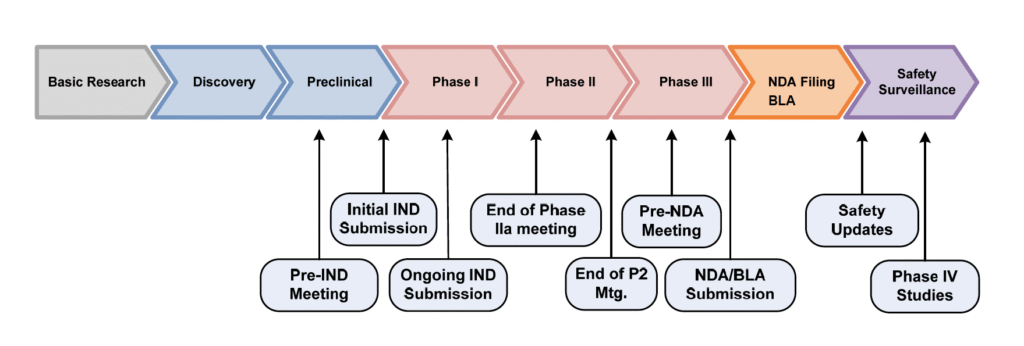
Pre-IND-möte – en introduktion av ett läkemedelsbolag för FDA
Ett pre-IND-möte är alltså ett möte med myndigheten som sker innan bolaget lämnar in sin IND-ansökan och är generellt den första kontakten mellan FDA och bolaget. Det är ett sätt för bolaget att presentera befintliga data, samt vilken data man ämnar samla in under studien, som kommer att utgöra basen för den planerade IND-ansökan.
Bolaget beskriver även de kliniska utvecklingsplanerna och får FDA:s återkoppling och rekommendationer inför den fortsatta kliniska utvecklingen. Detta säkerställer att myndigheten är eniga med bolagets planer och man undviker fördröjningar i den fortsatta processen.
»För RhoVacs del kommer pre-IND-mötet innebära att vi får insikt i FDA:s förväntningar på våra kliniska utvecklingsplaner. Därigenom ökar våra chanser att öppna en IND och beviljas tillstånd för en fas II-studie i ett senare skede.« – Ann Christine Korsgaard, Senior Director Regulatory Affairs på RhoVac
Flera beståndsdelar av ett pre-IND-möte
Exempel på ämnen som kan behandlas vid ett pre-IND-möte inkluderar:
- Om säkerhet i djur och människa dokumenteras på ett tillförlitligt sätt
- Om kemi, tillverkning och kontroll är tillräckligt dokumenterad för att produkten ska kunna användas i en klinisk studie
- Om FDA har några råd eller kommentarer till den föreslagna studiedesignen, t.ex. hur man mäter säkerhet och effekt, hur doseringen har fastställts m.m.
- Om kandidaten har potential att kvalificeras för något regulatoriskt snabbspår såsom Fast Track, Breakthrough Designation, Accelerated Approval eller Orphan Drug Designation
- Fast Track är en process utformad för att underlätta utvecklingen, och påskyndar granskningen, av läkemedel som utvecklas för att behandla allvarliga tillstånd och möta ett omättat medicinskt behov. Fast Track erbjuder mer frekventa kommunikationsmöjligheter med FDA och ökar chanserna för ytterligare incitament för snabbare regulatoriska processer.
- Breakthrough Designation är en process avsedd för att påskynda utvecklingen och granskningen av läkemedel som kan visa avsevärd förbättring jämfört med tillgängliga behandlingar och som redan har preliminära kliniska data som indikerar detta. Fördelarna liknar de för Fast Track, men med ännu mer vägledning från FDA.
- Accelerated Approval är ett sätt att tillåta läkemedel för allvarliga tillstånd som fyller ett omättat medicinskt behov att godkännas baserat på ett utfallsmått som inte är det finala utfallsmåttet. Med hjälp av ett tillfälliga utfallsmått kan FDA godkänna läkemedlet snabbare. Det tillfälliga utfallsmåttet måste på ett rimligt sätt förutsäga klinisk fördel även i det finala utfallsmåttet, såsom progressionsfri överlevnad istället för total överlevnad.
- Orphan Drug Designation ges till produkter som behandlar en sällsynt sjukdom. Incitamentet är ett specifikt marknadsskydd efter FDA-godkännande som motsvarar 7 års marknadsexklusivitet i USA.
Positivt utfall av likvärdig process med EMA

Vanligtvis hålls pre-IND-mötet innan bolagets första kliniska studie. Men då RhoVacs initiala studier genomförts i Europa har bolaget ansökt om mötet inför sin fas IIb. Pre-IND-processen hos FDA påminner dessutom mycket om den europeiska läkemedelsmyndigheten EMA:s procedur för vetenskaplig rådgivning – Scientific Advice – som RhoVac genomgick under våren 2018.
I samverkan med EMA konstaterades att inga ytterligare prekliniska studier behövdes för att stödja RhoVacs fas IIb-studie med RV001. EMA instämde dessutom i att det finns en tydlig behandlingsmöjlighet med RV001 i tidiga sjukdomsutvecklingen av prostatacancer.
Nästa steg i processen
När FDA nu bekräftat pre-IND-mötet har RhoVac kompletterar sin ansökan genom att skicka in ett Briefing Package. Myndigheten kommer nu att gå igenom materialet och lämna ett skriftligt utlåtande innan mötet, för att parterna sedan ska kunna hålla en givande och underbyggd diskussion vid det faktiska mötet.
BioStock fick en pratstund med Ann Christine Korsgaard som är Senior Director Regulatory Affairs på RhoVac och därmed ansvarig för att samordna bolagets regulatoriska aktiviteter.
Ann Christine Korsgaard, vill du börja med att berätta om din yrkesmässiga bakgrund?
– Efter en position hos SLK i Oslo 1992 insåg jag hur viktiga de regulatoriska frågorna är för läkemedelsutvecklande bolag. Sedan dess har jag lett ett antal internationella regulatoriska funktioner på biopharmabolag som Shionogi i Storbritannien, UCB i Belgien, Genmab, Action Pharma, Actavis, och LEO Pharma i Danmark.
– Jag har drivit globala regulatoriska processer för många projekt och guidat bolag genom strategiska, värdeskapande interaktioner med europeiska, amerikanska och japanska tillsynsmyndigheter. Detta inkluderar många pre-IND-möten med FDA.
– Min utbildningsbakgrund består av en doktorsexamen i farmakologi från Köpenhamns Universitet och en MBA från Scandinavian International Management Institute (SIMI).
»De ämnen vi kommer att diskutera med FDA är relaterade till utformningen av den planerade kliniska fas II-studien samt nivån av både icke-kliniska- och CMC-data, vilka krävs för att öppna en IND.«
Varför är det, enligt dig, viktigt för ett bolag som RhoVac att hålla ett pre-IND-möte med FDA och hur avgörande är utfallet av detsamma?
– FDA är en tillsynsmyndighet med betydande expertis som förväntar sig att bolagen engagerar dem i sina utvecklingsplaner i tid för att, vid behov, kunna justera planerna så att de matchar en möjlig regulatorisk rutt. För RhoVacs del kommer pre-IND-mötet innebära att vi får insikt i FDA:s förväntningar på våra kliniska utvecklingsplaner. Därigenom ökar våra chanser att öppna en IND och beviljas tillstånd för en fas II-studie i ett senare skede.
I ert pressmeddelande går att utläsa att ni i samband med mötesansökan också lämnade in specifika frågor som ni önskade att FDA skulle behandla. Kan du säga något om karaktären på dessa frågor?
– Ett pre-IND-möte är huvudsakligen avsett att vara ett tillfälle för att klargöra om tillgänglig data är lämpliga som underlag för att öppna en IND i USA för en särskild klinisk studie. De ämnen vi kommer att diskutera med FDA är relaterade till utformningen av den planerade kliniska fas II-studien samt nivån av både icke-kliniska- och CMC-data, vilka krävs för att öppna en IND.
»Vi har goda data från en europeisk first-in-man-studie med tydliga resultat gällande säkerhet och immunologiskt svar. […] Detta ger en mycket bättre position än när man har ett pre-IND-möte tidigare i utvecklingsprocessen och det inte finns någon klinisk data.«
Vad talar, i din mening, för att FDA ska vara lika positivt inställda till RhoVac och planerna för RV001 som EMA var våren 2018?
– RhoVac går in i diskussioner med FDA vid en tidpunkt då vi har goda data från en europeisk first-in-man-studie med tydliga resultat gällande säkerhet och immunologiskt svar. Detta gör det möjligt för FDA att granska vissa kliniska resultat samtidigt som man rådgör kring nästa förväntade steg mot den amerikanska marknaden. Detta ger en mycket bättre position än när man har ett pre-IND-möte tidigare i utvecklingsprocessen och det inte finns någon klinisk data.
– Därtill utvecklar RhoVac som riktar sig till patienter som behöver ett bra behandlingsalternativ där de kan bibehålla sin livskvalitet under behandlingen.
Vad händer om FDA:s utlåtande skiljer sig mycket från det RhoVac tidigare fått från EMA?
– Om FDA:s råd visar sig bli annorlunda än de vi tidigare fått från EMA så kan RhoVac överväga olika vägar framåt, t.ex.;
- Anpassa utformningen av fas II-studien genom att lägga fram ändringar i protokollet i Europa som tillgodoser FDA:s rekommendationer.
- Utarbeta en separat klinisk fas II-studie för USA som möter FDA:s rekommendationer.
Ni har nu skickat in ett s.k. Briefing Package till FDA. Vad kommer detta material att innehålla?
– Det är ett informationsdokument till FDA inför mötet som består av de sakfrågor vi vill stämma av med myndigheten, bakgrunden till varför och en bolagsposition som tydliggör vad vi anser att svaret borde vara. För varje sakfråga stämmer vi av om FDA håller med om vår tänkta strategi.
Ni väntar er FDA:s skriftliga svar på underlaget i slutet av april. Vad kommer detta svar att inkludera?
– FDA kommer att tillhandahålla skriftliga uttalanden till samtliga frågor vi har ställt till dem.
Vad händer efter pre-IND-mötet?
– Vi kommer att diskutera feedbacken från FDA internt på RhoVac och utvärdera om det är läge att gå vidare med en IND-ansökan i det skedet.
Ni har tidigare kommunicerat att fas IIb-studien ska inledas under mitten av 2019. Känns detta fortsatt som en rimlig tidsplan givet att pre-IND-mötet kommer att hållas i maj?
– Ja, vi kan under alla omständigheter starta studien i Europa för att sedan antingen anpassa protokollet i alla studie-länder genom en protokolländring eller göra en separat fas II-studie i USA.
Innehållet i Biostocks nyheter och analyser är oberoende men Biostocks verksamhet är i viss mån finansierad av bolag i branschen. Detta inlägg avser ett bolag som BioStock erhållit finansiering från. [et_bloom_inline optin_id=”optin_4″]
